

Summary
该协议描述了一种非侵入性方法,用于有效识别下游显微镜研究的S相细胞,例如通过激光微辐射测量DNA修复蛋白的招募。
Abstract
DNA损伤修复在高度反应的环境中保持细胞的遗传完整性。细胞可能由于内源和外源(如代谢活动或紫外线辐射)而积累各种类型的DNA损伤。如果不进行DNA修复,细胞的遗传密码就会受损,破坏蛋白质的结构和功能,并可能导致疾病。
了解不同细胞周期阶段不同DNA修复途径的间质动力学,对于DNA损伤修复领域至关重要。目前的荧光显微镜技术为测量DNA损伤诱导后不同修复蛋白的招募动力学提供了很好的工具。细胞周期S阶段的DNA合成是细胞命运中DNA修复的一个奇特点。它提供了一个独特的窗口来筛选整个基因组的错误。同时,DNA合成错误也对DNA完整性构成威胁,而非分裂细胞中是不会遇到的。因此,与细胞周期的其他阶段相比,S相的DNA修复过程存在显著差异,而且这些差异鲜为人知。
以下协议描述了使用配备 405 nm 激光线的激光扫描共聚焦显微镜,在局部诱导的 DNA 损伤部位 S 阶段的细胞系的制备和 DNA 修复蛋白动力学的测量。标记PCNA(带 mPlum)用作细胞循环标记,与感兴趣的 ACGFP 标记修复蛋白(即 EXO1b)相结合,用于测量 S 阶段的 DNA 损伤招募。
Introduction
一些DNA修复途径已经进化,以解决细胞中可能出现的不同类型的DNA病变,所有这些病变在空间和时间上都受到高度调节。细胞周期中最脆弱的时期之一是S相,当DNA合成发生时。虽然扩散对生命至关重要,但它也提供了一个重大挑战。细胞需要确保忠实地复制其基因组,以避免突变传给后代。因此,增殖提供了一个治疗干预点,用于在肿瘤学领域开发治疗方法。
所有用于研究DNA病变蛋白质招募的主要技术都有其优点和局限性。微辐照具有比大多数替代方法更好的空间和时间分辨率1, 如电离辐射诱发的foci(IRIF)、染色素免疫沉淀(ChIP)或生化分馏的免疫荧光成像。然而,微辐照带的坚固性上述技术,可以同时采样大量的细胞。
要研究S相的DNA修复,必须能够区分异步细胞培养群中的S相细胞。有许多众所周知的方法来解决这个问题,包括细胞的同步,或不同细胞周期阶段的可视化。然而,这两种方法都带来了重大挑战和可能的文物。化学同步方法广泛用于在早期S相丰富细胞(如双胸腺素块、阿菲迪科林和羟基尿素治疗),通过诱导复制应激并最终DNA损伤自身实现同步。这限制了使用这些方法研究DNA修复过程在S阶段2。通过血清饥饿和释放同步仅适用于数量有限的细胞系,主要不包括与未转化细胞系相比,对细胞周期进展依赖较少的癌细胞系。荧光育碧素细胞周期指标(FUCCI)系统是研究细胞周期的特别有用的工具,但在区分S和G2细胞周期阶段3时,它具有根本的局限性。
这里显示,使用荧光标记PCNA作为S相的非侵入性标记,可以限制化学细胞循环同步方法的缺点,同时允许比FUCCI系统更特殊性和灵活性。作为单个标记,PCNA 不仅可以突出显示异步种群中的 S 相细胞,而且还可以显示 S 相(即早期、中期或后期 S 相)4内细胞的确切进展。低表达水平的外源性,标记PCNA确保最小的干扰细胞周期的进展和DNA修复过程。重要的是,PCNA还作为适当的DNA损伤诱导的内部控制,因为它参与修复几个DNA病变,并被招募到当地诱导的DNA损伤点1,4。
这里介绍的实验演示了如何测量 EXO1b 在 S 阶段的招募动态,以及这如何受到成熟的 PARP 抑制剂 olaparib 的影响。EXO1b 核酸酶活性与广泛的 DNA 修复途径相关,包括不匹配修复 (MMR)、核苷酸切除修复 (NER) 和双链断裂 (DSB) 修复。在S阶段,EXO1b在DNA剖析5期间形成3'sDNA悬垂,在同源重组(HR)中起着重要作用。EXO1b进一步牵连到DNA复制与检查点激活的作用,重新启动停滞的DNA叉子,以及引金去除和冈崎碎片成熟在链位移过程中的链位移在复制5。EXO1b招募受损的DNA站点是由与多聚(ADP-核糖)(PAR)6,7的直接相互作用调节的。由于 EXO1b 具有众多细胞周期特定含义,因此对于使用 PCNA 进行 S 相特定招聘研究来说是一个绝佳的选择。
Protocol
1. 培养人类骨肉瘤衍生细胞(U-2操作系统)
注:U-2操作系统细胞非常适合这些研究,因为它们具有扁平的形态、大核和紧密连接到多个表面,包括玻璃。也可以使用具有类似特征的其他细胞系。
- 对于U-2操作系统细胞系的培养,使用麦考伊的5A介质补充10%胎儿牛血清(FBS)和抗生素(100U/mL青霉素和100微克/mL链霉素)。在含有 5% CO2的潮湿大气中在 37 °C 下孵育细胞。对于显微镜研究,保持细胞培养在10厘米的盘子,以提供足够的细胞计数。
- 当细胞接近90%的汇流(+7 x 106 细胞/10厘米盘),分裂细胞。
- 用PBS冲洗细胞,洗去血清中含有的试丁香抑制剂。
- 加入1兆L的Trypsin-EDTA,并确保细胞层被平等覆盖。
- 在 37 °C 下孵育,直到细胞层从板上抬起(约 6 分钟)。
- 将含有介质的血清中的试用细胞重新激活,使试剂素失活,并将体积的 1/10(+0.7 x 106细胞)添加到包含 10 mL 补充生长介质的新 10 厘米板中。
- 在实验之前,根据制造商的建议,使用通用支原体检测套件对细胞进行支原体污染的常规测试。
2. 逆转录病毒感染
注:有关 BSL-2 安全措施,在处理重组病毒时,请参阅:NIH 指南,III-D-3 节:组织培养中的重组病毒。
- 种子 4 x 106 HEK293T 细胞在电镀成 10 厘米培养皿后,可在 24 小时内实现 +60% 的汇流。
- 对于培养 HEK293T,请遵循本协议中 1.1-1.3 中描述的 U-2 操作系统的培养步骤。对于 Hek293t 替代麦考伊的 5a 媒体为 Dmem 。当 HEK293T 细胞弱弱地附着在组织培养板上时,请务必轻轻地清洗它们。
- 使用脂质转染试剂对质粒进行病毒包装的 HEK293T 细胞进行转化。
- 对于逆转录病毒载体,请将 1.5 μg 的 VSV-G(添加基因 #8454)和 1.5 μg 的 pUMVC(添加剂 #8449)包装载体相结合 以及含有感兴趣基因的3微克载体(在具有纯霉素耐药性的逆转录病毒载体骨干中),在微中心管中加入250微升Opti-MEM减少血清介质。加入Opti-MEM/DNA混合物(在此例中为6微升),并轻敲轻轻混合,每加入1微克P3000试剂。不要上下漩涡或移液器。
- 在另一个微中心管中,将每 μg DNA(在此例中为 12 μL)的转染试剂与 250 μL Opti-MEM 减少血清介质结合。
- 将两种混合物(500 μL 组合在一起,不要旋转,只需轻轻敲击混合),让它在室温下孵育 15 分钟。
- 小心地将混合物滴入种子的 HEK293T 细胞,而不会分离细胞。轻轻地旋转盘子。
- 病毒感染产生稳定的细胞系。
- 转染后 72 h 从 HEK293T 细胞中取出含有超高纳坦的病毒。使用 0.45 μm 过滤器仔细过滤溶液,以去除细胞碎屑和分离的细胞。可选,在病毒超高分子中加入 8 μg/mL 聚苯乙烯,以促进病毒感染。
- 将含有超纳坦的病毒添加到 U-2 操作系统细胞中,在 10 厘米盘(+3 x 106 细胞)中以 +50% 的汇流度添加。前一天播种 U-2 操作系统单元。
- 在去除和丢弃含有病毒的超高超纳特之前感染 6-16 小时。
注:为了达到对感兴趣的基因的预期过度表达量,在一定时间内培养一系列病毒稀释。检查每个新建立的细胞系中转基因的表达水平与西方斑点将其与内源水平进行比较。 - 允许细胞在适当的抗生素存在的情况下进行选择(在2μg/mL最终浓度的纯霉素的情况下3-4天),并在显微镜下验证荧光蛋白标记感兴趣的基因的表达。
- 重复这些步骤以生成双标记的细胞线。在本此间提出的实验中,mPlum-PCNA由逆转录病毒载体(pBABE)与EXO1B-ACGFP相结合,也从逆转录病毒载体(pRetroQ-ACGFP1-N1)表达。
3. 为微辐射制备细胞
- 电镀细胞:实验前24小时,将总共8.0×104 个细胞平板成500微升-1毫升介质(约70%的汇合)体积,在4个室室盖玻璃上,并配以1.5号玻硅酸盐玻璃底,为高放大镜和激光微辐射提供理想的结果。更高的细胞汇流允许在单个视场 (FOV) 中测量更多的细胞:然而,完全汇合的幻灯片将引入细胞周期不规则。
- 成像介质:在微辐照前一小时,用常规生长介质交换氟Brite DMEM,辅以10%FBS、100 U/mL青霉素和100微克/mL链霉素、15mM HEPES(pH=7.4)和1mM钠-丙烯酸酯。这种成像介质有助于最大限度地提高信号与噪声比,从而检测出非常暗的荧光。由于它包含 HEPES,因此在没有 5% CO2 大气的情况下,它也能稳定 pH 值。
- 在此步骤的成像前应用任何其他治疗。在下面提出的实验中,细胞在成像前一小时用奥拉帕里布(PARP抑制剂,最终浓度为1微米)或车辆控制(DMSO)1,8,9进行预处理。
4. 准备显微镜并选择S相细胞进行成像。
- 使用具有与此处概述的系统类似的属性的同焦系统,以获得最佳效果。这里提出的实验是使用安装在倒置显微镜支架上的共聚焦显微镜进行的(见 材料表)。
注:此处使用的显微镜配备了 50 mW 405 nm FRAP 激光模块和 60x 1.4 NA 石油计划-阿波克罗马特目标。共聚焦扫描头有两个扫描器选项:galvano 扫描仪(用于高分辨率)和共振扫描仪(用于高速成像)。- 通过软件控制的 XY galvano 设备将光出血 (FRAP) 激光后的荧光恢复引入样品。使用 488 nm 激光线激发 AcGFP 和 561 nm 或 594 nm 激光线来激发 mPlum。
注:以下滤光片组合提供最佳效果:使用 560 nm 长的通过过滤器,波长小于 560 nm 的发射光通过 ACGFP 的 525/50 nm 排放滤光片,而波长大于 560 nm 的发射光通过 595/50 nm 的排放滤波器传递。任何适当的过滤器集(例如,FITC/TRITC、GFP/mcherry、FITC/TxRed),确保最少的荧光流过可以使用。
- 通过软件控制的 XY galvano 设备将光出血 (FRAP) 激光后的荧光恢复引入样品。使用 488 nm 激光线激发 AcGFP 和 561 nm 或 594 nm 激光线来激发 mPlum。
- 打开环境室和显微镜组件。
- 在实验开始前至少 4 小时打开加热(舞台、目标和环境室)、CO2 供应和湿度调节器,以确保热平衡以获得稳定的图像采集。
- 在细胞转移到显微镜之前,至少 1 小时将光源与激光线一起初始化。
- 使用荧光标记的 PCNA 作为标记,在异步人群中选择 S 相细胞。按照下面的步骤做到这一点。
- 在 S 相中查找 mPlum 标记 PCNA 的独特本地化模式,从而可能识别此细胞周期阶段。PCNA 在细胞周期的 G1 和 G2 阶段的核中具有完全均匀的分布,同时被排除在细胞核之外。在 S 相中,PCNA 在核中重体的位置形成 foci。 图1 显示了整个S相的PCNA foci的不同模式,这使得甚至有可能区分早期、中期和后期S相。
- 透过眼部寻找一个FOV,它有足够的S相细胞进行微辐射。异步U-2操作系统细胞通常有30-40%的人口在S阶段。
- 尽量避免PCNA和感兴趣的蛋白质(POI)的表达水平(明亮和昏暗的细胞)的极端,在这种情况下EXO1b-ACGFP,这可能导致实验性人工制品。
- 找到合适的 FOV 时,尽量避免长时间扫描该字段,以尽量减少光出血和不需要的 DNA 损伤。
- 设置微辐照所需的感兴趣区域 (ROI)。使用相关软件(参见 材料表),首先插入二进制行(设置所需的行数和间距)设置所需的投资回报率。单击 二进制,然后单击 "插入"行|圆圈|椭圆形 绘制所需的线数。
- 将这些二进制行转换为 ROI,最后将这些 ROI 转换为刺激性投资回报率。为此,请首先单击 投资回报率,然后单击 "移动二进制"到 ROI,然后右键单击任何 ROI,然后选择 "用作刺激投资回报率:S1"。将这些线放入 FOV 中,以穿过细胞核。在整个协议中都使用了长度为 1024 像素的 ROI,该 ROV 横跨整个 FOV。
5. 免疫荧光染色或延时成像的微辐照。
- 确定最佳的微辐照设置。
- 在细胞进行微辐射之前,请拍摄 FOV 的更高分辨率图像,以识别 PCNA foci 以供以后分析。而不是连续扫描,同时记录使用的两个光通道(绿色和红色),以避免细胞在扫描之间的移动在两个波长。对于 foci 的正确分辨率,请使用至少 1024 x 1024 像素/场分辨率,1 倍变焦(此处使用的成像系统 0.29 μm 像素大小),扫描速度为 1/8 帧/s(4.85 μs/像素),平均为 2 倍。一旦这些参数设置在 A1 LFOV 紧凑型 GUI 和 A1 LFOV 扫描区域 窗口中,点击 捕获 按钮来记录 FOV。
注:在整个实验过程中保持相同的像素大小以确保可比结果非常重要。 - 要设置微辐照,请打开成像软件中的 ND 刺激 选项卡以访问 时间表(A1 LFOV /Galvano 设备) 窗口。这使用 galvano 扫描仪获取一系列刺激前图像,刺激(使用 LUN-F 50mW 405 nm FRAP 激光),然后使用 galvano 扫描仪再次获取一系列刺激后图像。首先在 时间安排 窗口中设置三个阶段。在 Acq/Stim 列中选择 收购|漂白| 分别为三个阶段进行收购。对于漂白阶段,将 S1 设置为投资回报率。
注:在此处介绍的实验中,在刺激阶段未获取任何图像。 - 在 Galvano XY窗口,设置了微辐照的关键因素:405纳米激光功率输出,停留时间(默认情况下迭代为1)。在此介绍的实验中,细胞以 100% 功率输出,以 1000-3000 μs 的居住时间,以 405 nm FRAP 激光照射(光纤尖端为 50 mW)。
注:由于激光居住时间是按像素计算的,只要像素大小保持不变,居住时间和功率密度之间的关系将在不同的FOV之间进行比较。 图2A 显示使用DNA损伤反应(DDR)通路特定蛋白质(DSB的FBXL10和NTHL1的氧化碱基损伤),以优化激光功率设置,以进行特定的损伤诱导。这些稳定的细胞系是在协议第2节之后产生的病毒感染。
- 在细胞进行微辐射之前,请拍摄 FOV 的更高分辨率图像,以识别 PCNA foci 以供以后分析。而不是连续扫描,同时记录使用的两个光通道(绿色和红色),以避免细胞在扫描之间的移动在两个波长。对于 foci 的正确分辨率,请使用至少 1024 x 1024 像素/场分辨率,1 倍变焦(此处使用的成像系统 0.29 μm 像素大小),扫描速度为 1/8 帧/s(4.85 μs/像素),平均为 2 倍。一旦这些参数设置在 A1 LFOV 紧凑型 GUI 和 A1 LFOV 扫描区域 窗口中,点击 捕获 按钮来记录 FOV。
- 时间推移成像。
- 使用时间 表、A1 LFOV 紧凑型 GUI 和 A1 LFOV 扫描区域 窗口为所需的时间窗口和间隔设置时间延迟成像。在此处介绍的实验中, EXO1b 和 PCNA 的招募被成像 12 分钟,每 5 秒扫描一次 FOV,扫描 1024 x 1024 像素/场,使用 1 倍变焦(导致此处使用的成像系统 0.29 μm 像素大小),扫描速度为 0.35 帧/秒(1.45 μs/像素),而平均而言,可减少照片漂白。
- 优化 激光功率百分比, 增益 和 偏移设置 ,以减少 在A1 LFOV紧凑型GUI 窗口成像过程中的光漂白。如果一个人的目标是测量POI和PCNA,使用同步扫描,而不是连续扫描,以避免细胞运动之间的扫描场为两个单独的荧光。
- 成像系统用于以下设置。对于 488 nm 激光线 (20 mW): 7% 激光功率, 增益: 45 (GaAsP 探测器) 与和偏移 2, 对于 561 nm 激光线 (20 mW): 4% 激光功率, 获得 40 (GaAsP 探测器) 与和偏移 2.
- 根据蛋白质的动能,延长或缩短图像之间的间隔或总时间间隔。在 时间表 窗口中,设置第三阶段获取行所需的 间隔 和 持续时间 。
- 现在按运行以执行微辐照和随后的时间推移成像。
- 在时间推移成像结束时,将刺激性 ROI 保存为单独的图像,这将有助于识别用于分析的任何下游软件中的微辐照坐标。
- 免疫荧光染色。
注:第5.1.3步和第 2A图 演示了使用已知的DNA修复蛋白来评估微辐照引入的DNA病变类型。修复细胞后,也可以使用特定的抗体检测某些DNA病变。也有可能通过内源性蛋白质的抗体检测来检测POI的招募。下图显示了检查 DSB 的 +H2A.X 的可视化 (图 2B)。 图3 显示了PCNA本地化和整个细胞周期对内源性和外源性标记PCNA的一致性。- 第 5.1.3 步之后,在微辐射后只拍摄一张图像,以确保基于 mPlum-PCNA 的招聘,进行适当的 FRAP 活动。注意 FOV 的确切坐标,以便在免疫荧光标记后找到该字段。
- 将细胞培养室从显微镜中取出,在 37 °C 的潮湿大气中孵育细胞,其中含有 5% CO2, 时间为 5-10 分钟。
注:副甲醛(PFA)是有毒的,工作应在通风良好的区域或烟气罩内进行。所有后续的清洗和孵化将在 4 井室滑梯中以 0.5 mL 的体积完成。孵化时间后,用 0.5 mL 的 PBS(137 mM NaCl、2.7 mM KCl、8 mM Na2HPO4和 2 mM KH2PO4)清洗细胞,并在室温下在 PBS 中用 0.5 mL 的 4% PFA 固定 10 分钟(RT)。 - 用 PBS 清洗细胞一次,然后用 50 mM NH4Cl 清洗它们,以淬火残留的 PFA。
- 在 Rt 用 Pbs 中的 0.1% Triton X - 100 使细胞渗透 15 分钟。
- 用阻塞缓冲器(5% FBS、3% BSA、PBS 中 0.05% 特里顿 X-100)将样品屏蔽 1 小时。
- 取出阻塞溶液,在 RT 的阻塞缓冲器中加入稀释的原发抗体(抗 +H2A.X,1:2000)1 小时。
- 用阻塞缓冲器清洗井 3 x 10 分钟。
- 在 RT 的阻塞缓冲 1 小时中加入稀释的二级抗体(抗鼠标 Alexa 488 Plus 结合体,1:2000)。
- 用阻塞缓冲器清洗井 3 x 10 分钟。
- 在 PBS 中使用 1 μg/mL DAPI 解决方案对核进行 15 分钟的计数。
- 用 PBS 清洗细胞一次。成像可直接在 PBS 或带有防褪料试剂(例如 AFR3)的 PBS 溶液中执行,以减少光出血。
6. 招聘分析
注:图4A显示Exo1b和PCNA招聘在DMSO或奥拉帕里布面前的代表性图像。图 4B显示用于数据分析的代表性图像。平均荧光值的计算方法是使用 mPlum-PCNA (A,黄色矩形) 在使用斐济的不同时间点上突出的激光轨道上使用矩形测量平均 ACGFP 强度。PCNA 可以用作内部控制,以突出显示沿投资回报率坐标的成功辐照。同样,平均 ACGFP 荧光值也用于计算核未损坏区域(B、蓝色矩形)。背景信号强度在无人居住区(C、红色矩形)测量,并从平均荧光值(图A和B)中减去。因此,每个数据收集点的相对平均荧光单元 (RFU) 由方程 RFU = (A + C)/ (B + C)8、9计算。由此产生的微辐照区域的 RFU 值在微辐照之前与 RFU 值正常化。
- 要定义微辐照位点的区域 A,则将细胞的核区、复制区和不规则核区域排除在测量之外。在斐济绘制两个 ROI 到将两个独立的区域为一个区域之间保持 移位 键。
注:蛋白质的招募因基因和辐照条件而异:因此,A区域的大小必须单独确定。一旦确定区域 A 的像素宽度,任何比较招聘都应保持不变。在此处介绍的实验中,使用了 7 像素宽矩形。 - 将录制视频期间移动的单元格排除在分析中。要包含高度移动的单元格,必须逐帧执行上述分析。
- 要可视化招聘配置文件,请使用统计软件绘制标准化的 RFU 值与时间对比。
- 使用曼-惠特尼测试计算 DMSO 和奥拉帕里布 (n+31) 治疗之间指示的时间点的差异。
Representative Results
细胞以特定的方式处理每种类型的DNA病变,这也取决于它们处于哪个细胞循环阶段。例如,在微辐射之后,双链断裂 (DSB) 将通过非同源端连接 (NHEJ) 或 HR 进行处理,具体取决于细胞周期阶段。在细胞周期的S和G2阶段,核素作用最广泛,产生DNA悬垂,这对适当的HR至关重要。为了促进S相细胞的评价,PCNA被用作单色细胞周期标记。 图 1A 显示了细胞周期过程中 mPlum-PCNA 的本地化配置文件。PCNA 在 G1 和 G2 阶段的核中具有完全均匀的分布(同时也大多被排除在核苷酸之外)。在 S 阶段,PCNA 定位到 DNA 复制点,可可视化为细胞核中的亮点。在早期S相细胞中,斑点相对较小,并且在整个细胞核中分布均匀。进入中S阶段,斑点变得模糊,并更向核和核的周界定位。在后期S阶段,点的数量减少,但变得越来越大,因为PCNA集中在后期复制点(图1B)。重要的是,pBABE矢量骨干的外源性PCNA表达低于内源水平,但足以通过显微镜检测,从而最大限度地减少细胞循环进展和DDR中的潜在人工制品。 图1C 显示PCNA过度表达的程度与内源水平相比。请注意,由于其尺寸较大,与 mPlum-PCNA 对应的频段迁移速度较慢。
我们的目标是在微辐射期间引入 DSB,以调查在 S 阶段对这些病变的 PARP1+2 依赖性招募 EXO1b。图2A显示,低剂量的能量(1000微米居住时间)不会诱发招募EGFP-FBXL10,DSB响应器(FRUCC复合物8的组成部分),而它足以诱导招募NTHL1-mCherry,一种基础切除修复(BER)通路蛋白,招募到氧化DNA损伤的部位10,11,12。在3000 μs的停留时间,EGFP-FBXL10和NTHL1-mCherry招募,展示了产生氧化病变和DSB的激光输出。 加强这些结果,图2B显示免疫荧光染色对\H2A.X (DSB标记),这显然是更明显的,当使用更高的能量剂量。PCNA 既是细胞循环标记,也是成功微辐照的标志,因为它通过激光居住时间设置充分招募人员。重要的是,外源和/或内源性荧光蛋白标记PCNA可用于本报记者的功能,因为他们的行为相似(图3)。内源性标记的 PCNA 是通过将 mRuby 插入框架中,将第一个外显子插入 PCNA 轨迹13的一个等位基因(细胞线是约格·曼斯菲尔德的一种礼物)来设计的。
图 4A和图 4C显示在 S 相单元中招募带有 ACGFP 标记的 EXO1b。EXO1b 在 1 分钟左右达到微辐照点的最大积累水平,然后慢慢开始脱离 DNA 病变。微辐射点的浓缩物由图表上> 1 个相对荧光单元表示。在奥拉帕里布的存在下,与车辆控制相比,1 分钟激光条纹上的 EXO1b 积累显著减少。这些结果与文献6,7是一致的。图 4B显示了协议第 6 点中描述的量化代表性区域(A 区、B 区和 C 区)。图4D显示了用于微辐照的细胞中内源性EXO1b和外源性EXO1b-ACGFP的可比表达水平。
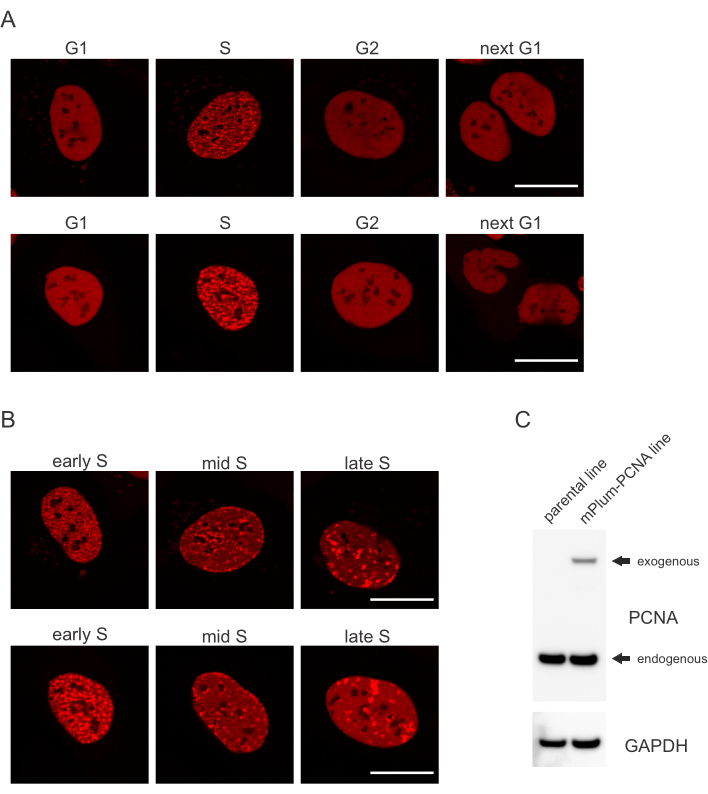
图1:PCNA的本地化模式。(A) 图像显示U-2操作系统细胞周期内稳定整合的外源PCNA的本地化模式。(B) 图像显示 U-2 操作单元中 S 相(早期、中期和后期)不同阶段的 PCNA foci 模式。(C) 用于成像的U-2操作系统细胞中显示PCNA内源性和外源性水平的西性污点。缩放条表示 20 μm。请单击此处查看此图的较大版本。

图2:通过优化的激光功率输出诱导DSB。 (A) 可以优化激光设置,以诱导不同形式的DNA损伤。U-2操作系统细胞稳定地表达EGFP-FBXL10和NTHL1-mCherry分别用于识别DSB和氧化病变部位。在具有 1000 μs 或 3000 μs 的异步 U-2 操作系统单元上使用 405 nm 激光线进行微辐射。比例杆代表 20μm.(B)免疫荧光染色对 \H2A.X 是在人类视网膜色素上皮细胞 (hTERT RPE-1) 有 mRuby 标记的内源性 PCNA 上完成的。细胞在微辐射后5分钟得到修复和处理,停留时间为1000微米或3000微米。缩放条表示 20 μm。 请单击此处查看此图的较大版本。

图3:可比较地将内源性mRuby-PCNA和外源性mPlum-PCNA以1000微米或3000微米的激光居住时间招募到微辐射场。 内源性与外源性标记 PCNA 在 S 相期间形成复制 foci。 请单击此处查看此图的较大版本。

图4: PARP1+2 依赖在 S 阶段招募 EXO1b。 U-2 操作系统单元稳定地表达 EXO1b-AcGFP 和 mPlum-PCNA 使用 3000 μs 的停留时间用 405 nm FRAP 激光线进行微辐射。(A) 车辆控制 (DMSO) 或奥拉帕里布 (1 μM) 进行预处理后,在指示的时间点对微辐照细胞的代表性图像。比例表表示招聘分析中 A、B 和 C 区域的 20 μm.(B)代表图像。比例条表示 20 μm.(C)DNA 损伤招募动态被活细胞成像捕获。相对平均荧光值和图像每 5 秒获得 12 分钟。对于每个情况,≥30个细胞进行了评估。平均相对荧光值(实心黑线)和标准误差(按阴影区域可视化的范围)是根据时间绘制的。虚线显示微辐照后 1 分钟的招聘值。DMSO (n=32) 和奥拉帕里布 (n=31) 治疗之间的区别是使用曼-惠特尼测试计算的。星号表示 p <0.0001。(D) 西方污点比较用于微辐照的细胞中内源性EXO1b和外源性EXO1b-ACGFP的表达水平。 请单击此处查看此图的较大版本。
Discussion
关键步骤和潜在协议故障排除/修改
用于微辐照的适当组织培养容器对成功至关重要。大多数高分辨率成像系统都优化为 0.17 mm 盖玻璃厚度。使用厚度较高或更低的成像室或塑料聚合物制成的成像室(未优化为 405 nm 成像),可显著降低图像质量。使用玻璃表面时,确保它们经过组织培养处理,以增强细胞粘附性。如果他们没有组织培养治疗,这些腔室将需要涂层,例如,在播种细胞之前,用多D-赖氨酸。当将细胞电镀到室内盖玻璃中时,理想的细胞密度对于避免细胞周期的不规则性和细胞的额外压力至关重要。在实验前,显微镜组件的适当热平衡以保持稳定的温度对于在整个时间差成像过程中保持对焦至关重要,并且对于确保跨时间和样品的均匀 DDR 也是必要的。
在微辐照之前,细胞处于健康状态以减少人工数据至关重要。如果细胞在感染/选择后有不规则的形态,允许细胞通过多个通道前进,直到形态恢复正常。始终确保使用的细胞线不受支原体污染。在支原体感染的许多不利影响中,它也对宿主细胞造成DNA损伤,并可能影响其DDR通路14,15。检测细胞培养中支原体的最敏感方法是通过 PCR(与 DAPI 或 Hoechst 的检测相比)。
然而,对感兴趣的修复蛋白的最佳过度表达应可与内源水平相媲美,其含量足以检测。病毒载体上使用的促进剂、感染期间的病毒滴答声和感染时间长度都可以根据理想的表达水平进行调整。为了获得一致的结果,隔离单个细胞克隆,以确保均匀的表达水平和正常的细胞形态。建议使用不过度表达标记 PCNA 在高于内源水平的矢量结构,以获得适当的细胞循环和 DNA 修复标记功能。即使是低水平的PCNA过度表达也足以区分S相细胞。逆转录病毒 pBABE 载体已成功用于此目的(添加基因 #1764,#1765,#1766,#1767)。PCNA 可以标记为任何单体红色(如 mPlum、mCherry、mRuby 等)或单体绿色荧光蛋白(例如,mEGFP、ACGFP、mWasabi、mNeonGreen、mEmerald 等),然后可以与交替标记的 POI 组合。过度表达荧光标记的 POI 有一些限制和考虑。荧光标签可能会破坏正常的蛋白质功能和本地化。因此,必须考虑标签的位置(N 或 C 终端)。始终使用单体荧光蛋白,因为非单体变异的寡聚会影响 POI 的功能。
必须为每个成像系统确定激光设置,因为光学路径的许多组件将影响输送到细胞的实际功率。激光微辐照可根据激发波长、FRAP 激光的功率输出以及是否使用任何预敏剂(如溴二氧尿素或霍奇斯特)导致多种类型的 DNA 病变。405纳米激光可引起氧化DNA损伤,单双搁浅断裂16,17。通过使用更高的激光输出设置,DSB 的数量会增加。在本协议中,没有使用预敏方法,但这些技术在文献中得到了很大的涵盖,并在下面的讨论中重新盖上盖。我们认为,测试所需病变是否产生的最好方法是检测已知DNA损伤途径特定基因的招募情况。招募NTHL1或OGG1,BER通路的组成部分,表明氧化DNA碱基的诱导10,11,17,18,19,而FBXL10或XRCC5表明DSB的存在8,20,21。XRCC1的招募可以表明氧化DNA碱基和单搁浅断裂(SSB)22,23的存在。XPC(即RAD4)是NER的一个很好的指标,它去除紫外线(UV)17,24产生的笨重的DNA导管。由于招募外源性蛋白质可能会引入某些不规则性,内源性DNA修复蛋白或标记物(如双搁浅断裂的 +H2A.X)的免疫荧光染色可以确认特定 DNA 病变的存在。或者,也可以使用针对特定类型的DNA病变而提出的抗体。为了调整交付的激光功率,可以更改居住时间和激光功率。
在数学建模的帮助下,可以进行详细的动能分析,从而对 POI 的招募特性(例如,多个 DNA 结合域的贡献、对不同信号事件的敏感性等)提供有价值的见解。自动招聘评估和细胞跟踪可以结合起来,创造强大的工作流程1,25。
DNA预敏化的优势和局限性
微辐照前对DNA进行预敏是DNA修复蛋白质招募的常用工具。在微辐射之前对DNA进行敏感,使其更容易受到DSB的影响。DNA预敏化的两种最常见的方法是用溴氧尿素(BrdU)或霍奇斯特染料对细胞进行预处理。对于不能在高激光功率下进行微辐射的系统,这些方法对于诱导DNA病变(如DSB)可能是必要的。 此外,在没有显示细胞核的传输光探测器或荧光信号的情况下(例如,在研究未标记的内源性DNA修复蛋白的招募时),Hoechst既是一种预敏工具,也是一种荧光核污渍。然而,DNA预敏化可以带来严重的并发症。BrdU(在最终浓度为10μM时使用)必须24小时(或相当于使用细胞系中的整个细胞周期的时间)添加到细胞中,以正确地融入DNA,并可能导致细胞周期干扰26。Hoechst 33342(最终浓度为1微克/兆升)在经过长时间的潜伏期后具有细胞毒性,但需要足够的时间用染料使细胞核饱和。因此,它只应在微辐射前15-20分钟应用:否则,招聘数据将不一致。这种方式染色的细胞不能保存在培养超过几个小时27,28。请务必不要使用霍奇斯特 33358,它不像霍奇斯特 33342 染料那样具有细胞渗透性。预敏化还可以在实验之间引入不必要的差异,使实验对细胞密度的差异更加敏感(因为这将影响结合染料/细胞的数量)。
共聚焦显微镜的优势和局限性
与广域显微镜相比,共聚焦显微镜的成像速度可能会受到限制。然而,配备共振扫描仪的共聚焦显微镜可以极大地提高成像速度(以分辨率为代价),接近旋转盘显微镜的速度。三个功能使 A1R HD25 通信系统成为此处介绍的协议的绝佳选择。首先,系统的 25 mm FOV 使在单个扫描场中对 15-20 个单元格进行成像(在常规设置中为 5-10 个单元格)中能够进行成像,从而限制了获取足够的细胞进行统计分析所需的获取次数。其次,FRAP模块和两个扫描头可以同时对细胞进行成像和微辐射,而不仅仅是按顺序进行。最后,具有谐振和 galvano 扫描仪的灵活性提供了以极快的速度轻松切换高时态分辨率成像和利用较慢的扫描速度生成信号与噪声比的高空间分辨率成像之间的切换能力。虽然使用的系统允许上述灵活性,类似于更广泛可用的共聚焦显微镜配置,但只有galvano扫描仪用于所呈现的实验(用于微辐照和后续成像)。
微辐照的优势和局限性
虽然微辐射提供了无与伦比的空间和时间分辨率,但它并非没有限制。与自然产生的损害剂相比,激光微辐射对细胞核特定部位的DNA损伤高度聚类。因此,微辐照产生的染色质反应可能与同质分布损伤不同。此外,微辐射是耗时的,可能只对几十个细胞进行,而大型基于人群的生化方法(色质分馏、免疫沉淀、ChIP)可以通过一次研究数千个细胞来增强健壮度。用传统的生化技术验证微辐射观测结果是得出可靠结论的有效策略。虽然在某个 FOV 中同时对许多细胞进行微辐射是可能的,但成像系统将需要更多的时间来执行任务。因此,测量对DNA病变迅速招募的蛋白质的动态限制了同时用于微辐照的可能ROI的数量。在用于此协议的成像系统上,单个 1024 像素长的投资回报率的微辐照需要 1032 ms 使用 1000 μs 居住时间,使用 3088 ms 使用 3000 μs 居住时间完成。使用多行 ROI 将显著增加完成微辐照所需的时间(例如,7 x 1024 像素长的投资回报率需要 14402 ms 使用 1000 μs 居住时间,21598 ms 使用 3000 μs 居住时间)。这一次因图像采集而丢失,必须加以考虑。在成像快速招聘事件时,尽可能使用最短的投资回报率,并且一次只对一个单元格进行微辐射。
与同步方法相比的优势和局限性
对于细胞周期的特定研究,现有方法包括细胞同步到特定的细胞周期阶段,或者使用荧光记者来识别细胞的特定细胞周期阶段。然而,每种方法都提供了自己的挑战和局限性。
FUCCI系统3( 依靠荧光蛋白标记截断形式的CDT1和Geminin)是细胞周期研究的特别有用的工具,但在区分细胞周期的S和G2阶段时有局限性。Geminin 水平已经从 S 中阶段高,并保持高到 M 相,使这些阶段难以分离。使用 FUCCI 系统还意味着显微镜的两个光学通道不能用于成像 POI。
非癌细胞系可以通过去除血清(血清饥饿)中的生长因子同步到G0,对细胞造成很少或根本没有DNA损伤。然而,大多数癌细胞系将部分继续通过细胞周期的进展,即使没有足够的血清在其介质。此外,细胞部分开始失去同步后期G1,早期S阶段。除了血清饥饿,还有许多化学方法实现细胞周期同步。羟基尿素、阿菲迪科林和胸腺素块是阻止DNA复制的方法,可将细胞同步到早期S相。虽然这些方法便宜而简单,但它们引入了复制应力,导致DNA损伤。这些DNA复制抑制剂已被证明能诱导H2A的磷化。X,DSB2,29的著名标记。使用标记-PCNA作为S相细胞标记的方法降低了化学同步引起的人工制品的可能性,并且与血清缺乏相比,可以应用于广泛的细胞系。
结论
DNA损伤是遗传性疾病的驱动力,诱因性病变可导致细胞的恶性转化。以DNA合成机制为目标,是治疗癌症等高扩散性疾病的基本治疗策略。为了更有针对性地治疗这些疾病,我们需要更好地了解修复DNA病变的蛋白质。此处描述的协议通过最大限度地减少传统同步方法带来的挑战,减少可能的人工制品并增加实验的可重复性,帮助 S 阶段的微辐照研究。
Disclosures
作者指出,所展示作品的出版是由尼康公司赞助的。作者宣称不存在相互竞争的利益。
Acknowledgments
作者感谢帕加诺先生对手稿的不断支持,以及西蒙内斯基、阿·马齐奥和唐大夫对手稿的批评性评论。B. 米瓦塔尼-明特感谢米瓦塔尼和布·明特的继续支持。罗娜感谢罗纳·侏罗兹和罗娜的继续支持。
Materials
| Name | Company | Catalog Number | Comments |
| Ammonium chloride | Sigma-Aldrich | A9434-500G | For quenching formaldehyde |
| Anti-EXO1 Rabbit Polyclonal Antibody | Proteintech | 16253-1-AP | primary antibody |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | 05-636 | primary antibody |
| Bovine Serum Albumin | Sigma-Aldrich | 3117332001 | BSA for blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Merck | 19-160 | pre-sensitizing agent |
| Citifluor™ Mountant Solution AFR3 | Electron Microscopy Sciences | 17973-10 | antifade containing PBS solution for imaging |
| DAPI | Sigma-Aldrich | D9542-1MG | nucleic acid stain |
| DMEM Medium | Thermo Fisher Scientific | 10569010 | Cell culture medium for HEK293T cells |
| DMSO | Sigma-Aldrich | D2650-100ML | Vehichle control and dissolution solvent |
| EGFP-FBXL10 | Addgene | #126542 | viral expression vector for EGFP-FBXL10 |
| EXO1b-AcGFP (in pRetroQ) | custom cloning | na | EXO1b cDNA was cloned in the NheI, BamHI sites of pRetroQ-AcGFP1-N1 vector. |
| Fetal Bovine Serum | Gibco | 16140071 | Media supplement |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 | Phenol red free medium for microscopy |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A32723 | secondary antibody |
| HEK293T cells | ATCC | ATCC CRL-3216 | Cell line for viral packaging |
| HEPES | Sigma-Aldrich | H0887-100ML | Buffering agent to supplement live cell imaging medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | pre-sensitizing agent |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 | Transfection reagent |
| McCoy’s 5A (Modified) Medium | Life Technologies | 16600-108 | Cell culture medium for U-2 OS cells |
| mCherry-PCNA | Addgene | #55117 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA | Addgene | #55994 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA (in pBABE) | custom cloning | na | mPlum-PCNA cDNA was cloned from Addgene #55994 in the BamHI, SalI sites of pBABE (puro) |
| Nikon A1R-HD25 Confocal Scanhead and Controller | Nikon | na | confocal imaging system |
| Nikon LUN4 laser unit | Nikon | na | excitation system |
| Nikon LUN-F 50 mW 405 nm FRAP laser unit | Nikon | na | FRAP laser unit |
| Nikon NIS Elements Confocal Controller Software | Nikon | na | Confocal controlling software |
| Nikon Ti2-E Inverted Microscope | Nikon | na | inverted epifluorescent microscope base |
| Nikon Ti2-LAPP Modular Illumination System | Nikon | na | illumination system |
| NTHL1-mCherry (in pRetroQ) | custom cloning | na | NTHL1 cDNA was cloned in the NheI, SalI sites of pRetroQ-mCherry-N1 vector. |
| Nunc Lab-Tek II Chambered Coverglass (4 well) | Thermo Fisher Scientific | 155382PK | Live cell microscopy cell culture chamber |
| Olaparib | Selleck Chemicals | S1060 | PARP inhibitor |
| Opti-MEM reduced serum media | Thermo Fisher Scientific | 31985062 | Dilution medium for transient transfection |
| Paraformaldehyde aqueous solution (32%) | Thermo Fisher Scientific | 50-980-494 | Fixative |
| pBABE (hygro) | Addgene | #1765 | retroviral expression vector (for low expression levels) |
| pBABE (neo) | Addgene | #1767 | retroviral expression vector (for low expression levels) |
| pBABE (puro) | Addgene | #1764 | retroviral expression vector (for low expression levels) |
| pBABE (zeo) | Addgene | #1766 | retroviral expression vector (for low expression levels) |
| PCNA Antibody (PC10) | Santa Cruz | sc-56 | primary antibody |
| Penicillin-Streptomycin-Glutamine (100x) | Gibco | 10378016 | Media supplement |
| polybrene | Sigma-Aldrich | TR-1003 | Increase viral infection efficiency |
| pRetroQ-AcGFP-C1 | Takara | 632506 | retroviral expression vector |
| pRetroQ-AcGFP-N1 | Takara | 632505 | retroviral expression vector |
| pRetroQ-mCherry-C1 | Takara | 632567 | retroviral expression vector |
| pRetroQ-mCherry-N1 | Takara | 632568 | retroviral expression vector |
| pUMVC | Addgene | #8449 | Viral packaging vector |
| Sodium-pyruvate | Thermo Fisher Scientific | 11360070 | Supplement for live cell imaging medium |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | Dilute in PBS for cell permeabilization buffer |
| Trypsin-EDTA Solution 10X | Sigma-Aldrich | 59418C-100ML | Dilute in PBS to split cells |
| U-2 OS Cells | ATCC | HTB-96 | Optimal cell line for microscopy experiments |
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K | PCR based Mycoplasma detection kit |
| VSV-G | Addgene | #8454 | Viral protein envelope vector |
References
- Aleksandrov, R., et al.
- Darzynkiewicz, Z., Halicka, H. D., Zhao, H., Podhorecka, M. Cell synchronization by inhibitors of DNA replication induces replication stress and DNA damage response: Analysis by flow cytometry. Methods in Molecular Biology. 761, 85-96 (2011).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Herce, H. D., Rajan, M., Lattig-Tunnemann, G., Fillies, M., Cardoso, M. C. A novel cell permeable DNA replication and repair marker. Nucleus. 5 (6), 590-600 (2014).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in dna replication with putative roles in cancer. International Journal of Molecular Sciences. 20 (1), (2018).
- Cheruiyot, A., et al. Poly(ADP-ribose)-binding promotes Exo1 damage recruitment and suppresses its nuclease activities. DNA Repair (Amsterdam). 35, 106-115 (2015).
- Zhang, F., Shi, J., Chen, S. H., Bian, C., Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Research. 43 (22), 10782-10794 (2015).
- Rona, G., et al. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. elife. 7, (2018).
- Young, L. M., et al. TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the dna damage response. Cell Reports. 13 (3), 451-459 (2015).
- Kong, X., et al. Laser microirradiation to study in vivo cellular responses to simple and complex dna damage. Journal of Visualized Experiments. (131), e56213 (2018).
- Kong, X., et al. Condensin I recruitment to base damage-enriched DNA lesions is modulated by PARP1. PLoS One. 6 (8), 23548 (2011).
- Lan, L., et al. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Research. 42 (4), 2330-2345 (2014).
- Zerjatke, T., et al. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Reports. 19 (9), 1953-1966 (2017).
- Ji, Y., Karbaschi, M., Cooke, M. S. Mycoplasma infection of cultured cells induces oxidative stress and attenuates cellular base excision repair activity. Mutation Research. 845, 403054 (2019).
- Sun, G., et al. Mycoplasma pneumoniae infection induces reactive oxygen species and DNA damage in A549 human lung carcinoma cells. Infection and Immunity. 76 (10), 4405-4413 (2008).
- Gassman, N. R., Wilson, S. H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair (Amsterdam). 31, 52-63 (2015).
- Muster, B., Rapp, A., Cardoso, M. C. Systematic analysis of DNA damage induction and DNA repair pathway activation by continuous wave visible light laser micro-irradiation. AIMS Genetics. 4 (1), 47-68 (2017).
- Ikeda, S., et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. Journal of Biological Chemistry. 273 (34), 21585-21593 (1998).
- Rosenquist, T. A., Zharkov, D. O., Grollman, A. P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proceedings of the National Academy of Science U. S. A. 94 (14), 7429-7434 (1997).
- Reid, D. A., et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Science U. S. A. 112 (20), 2575-2584 (2015).
- Taccioli, G. E., et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 265 (5177), 1442-1445 (1994).
- Marsin, S., et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. Journal of Biological Chemistry. 278 (45), 44068-44074 (2003).
- Thompson, L. H., Brookman, K. W., Jones, N. J., Allen, S. A., Carrano, A. V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Molecular and Cell Biology. 10 (12), 6160-6171 (1990).
- Scharer, O. D.
- Oeck, S., et al. High-throughput evaluation of protein migration and localization after laser micro-irradiation. Science Reports. 9 (1), 3148 (2019).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Science Reports. 6, 19567 (2016).
- Durand, R. E., Olive, P. L. Cytotoxicity, mutagenicity and dna damage by hoechst 33342. Journal of Histochemistry and Cytochemistry. 30 (2), 111-116 (1982).
- Tobey, R. A., Oishi, N., Crissman, H. A. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proceedings of the National Academy of Science U. S. A. 87 (13), 5104-5108 (1990).
- Podhorecka, M., Skladanowski, A., Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. Journal of Nucleic Acids. 2010, (2010).
